Overview: Tumor signaling systems
The Blumer lab studies signal transduction in tumors. We focus on answering the most conceptually and therapeutically important questions by revealing novel signaling networks that drive tumors and determining how these mechanisms can be targeted for therapeutic development. Our multidisciplinary research gives students and postdocs the opportunity of exploring ideas over a wide range of physical and biological scales, including at the level of single molecules, cells, omics, mouse models, and patient tumors. Trainees from the lab have gone on to enjoy successful scientific careers in academics and industry (see People).
For information about some of our current projects, click on the headings below.
Oncogenic guanine nucleotide-binding proteins such as Ras and G protein α-subunits are expressed in nearly half of all human tumors, potentially making them targets for therapeutic development. However, oncogenic G proteins have long been considered to be “undruggable” because their high affinity for GTP has precluded development of inhibitors that compete for nucleotide binding.
We developed an alternative approach by showing that an oncogenic G protein α-subunit can be inhibited allosterically. We demonstrated that allosteric inhibitors (YM-254890 or FR900359 (YM/FR)) can trap the inactive GDP-bound state of an oncogenic G protein α-subunit (Fig below). As a result, YM/FR shuts off all downstream oncogenic signaling networks, arrests tumor cell proliferation, and stops growth of xenografted tumors.
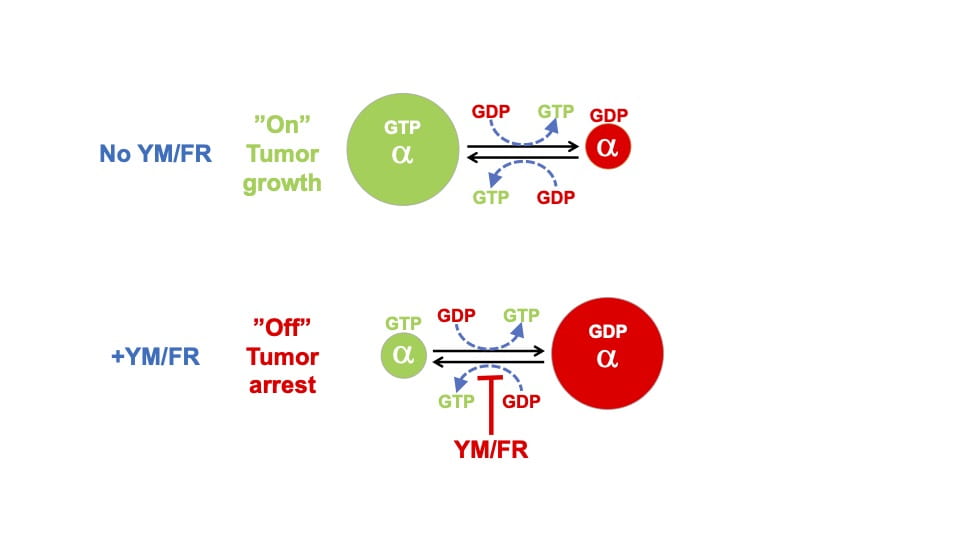
Our current studies use systems biology approaches to elucidate downstream oncogenic signaling mechanisms that drive critical processes in tumors and provide additional targetable vulnerabilities for therapeutic development.
How YM/FR allosterically inhibit their G protein α-subunit targets has remained unknown for more than a decade. This question must be answered in order to design YM/FR analogs that target other classes of G protein α-subunits and thereby provide novel experimental therapeutics for a wide range of diseases. The key question is how YM/FR inhibits GDP release. We recently have answered this question by using single-molecule FRET and molecular dynamics simulations to show that YM/FR interferes with an event required for GDP release: a clamshell-like opening between the Ras-like and helical domains of the Gα subunit (Fig below).
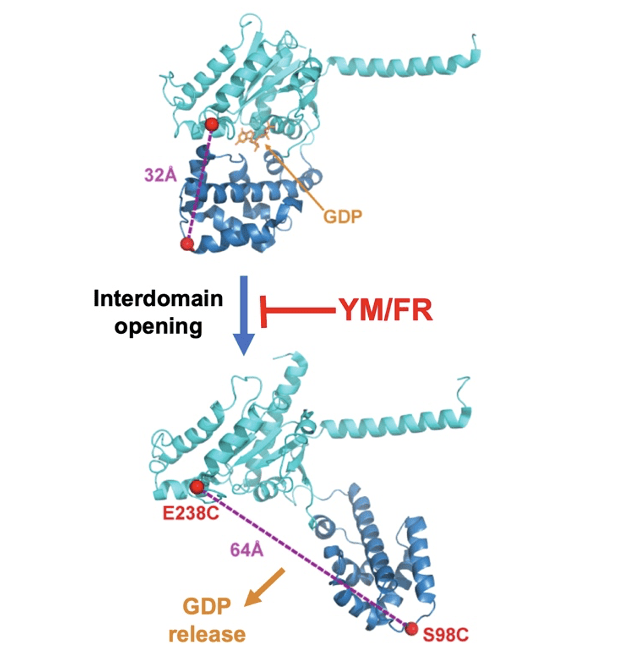
We also showed this mechanism is universal because all mammalian Gα subunits possess homologous but degenerate YM/FR binding sites yet can be inhibited by YM/FR upon introduction of an optimized YM/FR binding site.
With this new knowledge, our ongoing research seeks to design, synthesize, and characterize inhibitors tailored to each class of G protein α-subunit, thereby providing powerful new tools to probe G protein function and develop therapeutic agents for a broad spectrum of diseases. We are pursuing these goals by using in silico docking, synthetic organic chemistry, biochemistry, cell signaling, and physiological assays.
Tumor cells can survive inhibition of oncogenic G protein signaling by inducing adaptive survival mechanisms. To identify and target these survival mechanisms, we are using systems biology approaches, including synthetic lethal screens and computational network analysis of multi-omics data. For example, our network analysis of tumor phosphoproteomic data has shown that YM/FR arrests cells by inactivating cyclin-CDK complexes required for cell cycle progression (Fig below).
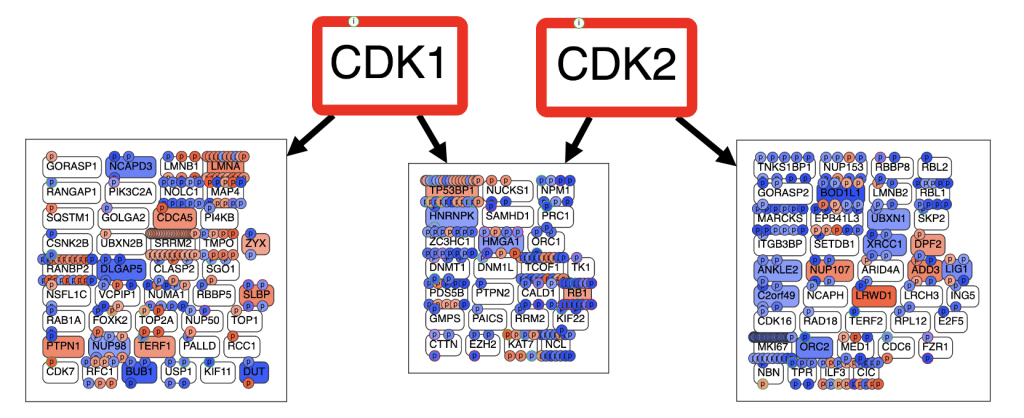
Ongoing synthetic lethal screens and network analysis of multi-omics data are providing a wealth of novel targets revealing new biological regulatory mechanisms in tumors that can be targeted combinatorially for therapeutic investigation.